
分子动力学模拟是一种利用计算机模拟技术研究分子系统物理性质的方法,主要基于牛顿运动方程来模拟分子体系的运动,下面将深入分析GROMACS软件、计算化学的硬件需求、分子动力学模拟的基本过程、关键技术GPU加速以及模拟信息处理与数据分析:

1、GROMACS软件应用
软件概述:GROMACS是一款用于进行分子动力学模拟的软件,广泛应用于生物化学分子的研究,如蛋白质、脂质和核酸等。
系统兼容:GROMACS能够模拟具有数百万颗粒子的系统,满足大规模分子动力学模拟的需要。
模拟原理:它基于牛顿运动方程对分子行为进行模拟,允许科学家研究在给定条件下的分子互动和动态行为。
应用领域:主要用于生物大分子的模拟,但其也可用于其他复杂键合相互作用的分子系统分析。
广泛使用:由于其强大的功能和用户友好性,GROMACS成为分子动力学模拟的首选工具之一。
2、计算化学硬件需求
预算考虑:进行高效的分子动力学模拟需要一定的硬件预算,如16.5万的预算可以通过学校招标方式购置相应设备。
性能要求:对于分子动力学模拟和分子对接这类高性能计算任务,推荐使用支持GPU加速的计算系统。
性价比:使用中档GPU就能达到甚至超过使用传统双路服务器的速度,因此具有很高的性价比。
长远规划:除了当前的主要功能,考虑到未来可能的基因组数据分析等应用,选择可扩展的硬件平台是明智的选择。
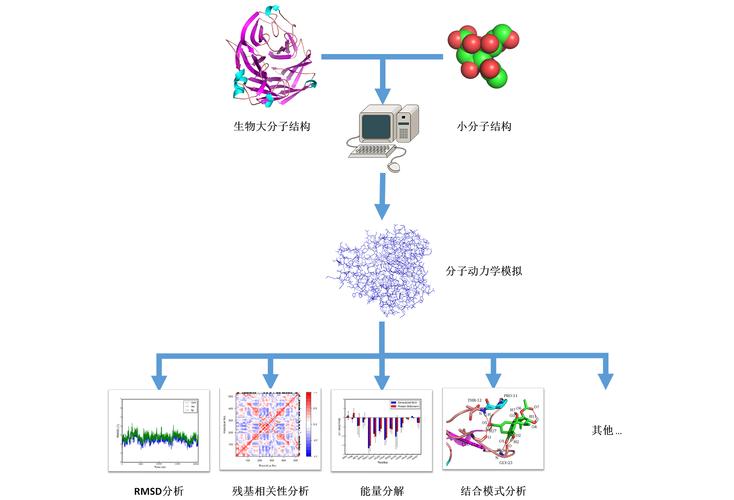
3、分子动力学模拟过程
模拟初始化:需要读取体系中分子的三维坐标、速度和粒子数密度等参数作为初始条件。
参数选择:选取合适的时间步长和势函数,这对于确保模拟精度和效率至关重要。
力和运动计算:模拟过程中需不断计算分子受到的作用力和加速度,并更新分子的速度和位置。
轨迹获取:重复上述计算过程,获得分子动力轨迹,由此得到宏观物理参数,并与实验数据进行对照。
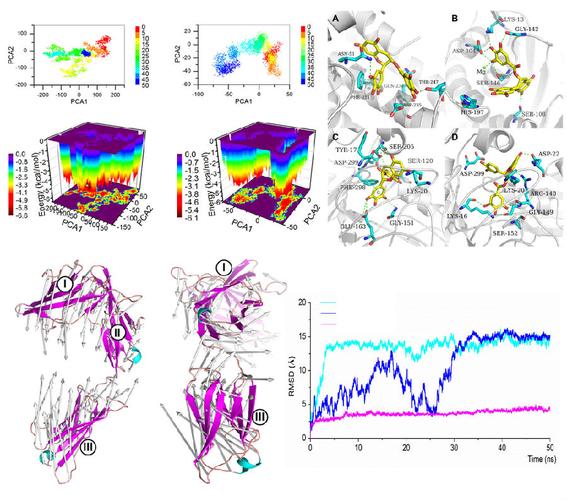
4、GPU加速技术
并行计算优势:在分子动力学模拟中,力的计算是并行的,各个分子受力计算相互独立,因此适合采用GPU加速。
程序支持:GROMACS、AMBER、NAMD和Desmond等分子动力学程序都支持GPU加速,充分利用现代GPU的高计算能力。
性能提升:使用GPU加速可以显著提高计算效率,使得模拟过程更快,从而节省时间和计算资源。
5、数据分析过程
轨迹分析:通过得到的分子动力轨迹可以进行深入的数据分析,以了解分子水平上的行为和反应。
宏观参数:模拟结果可以与实验数据进行比较,验证模型的准确性,并为实验设计提供理论依据。
在深入了解分子动力学模拟的基础上,还需考虑一些关键的操作和实践建议,以确保模拟的成功和高效:
准确建模:在模拟前要仔细检查和验证所选模型的准确性,保证模拟结果的可靠性。
调试测试:进行初步的试运行和调试,以确认参数设置的合理性和系统稳定性。
结果验证:模拟得到的结果需要进行详尽的验证,必要时与实验数据对比,以评估其科学性。
持续监控:对于长时间的模拟,需要监控系统运行状态,确保计算资源的有效利用和模拟进程的平稳运行。
归纳而言,分子动力学模拟是一个复杂的计算过程,涉及众多科学计算和模型分析,GROMACS等软件为此类模拟提供了强大的工具,而配备适当的硬件,特别是支持GPU加速的系统,可以极大提高模拟的效率和效果,在进行分子动力学模拟时,必须仔细考虑各种参数的选择,确保模拟的精确度,并且对得到的数据进行详尽的分析,以得出有意义的科学上文归纳。
【版权声明】:本站所有内容均来自网络,若无意侵犯到您的权利,请及时与我们联系将尽快删除相关内容!


发表回复